AG Schweickert
Forschung:
Wir untersuchen molekulare Mechanismen der Embryonalentwicklung. Unser wichtigster Modellorganismus ist der Krallenfrosch Xenopus, in geringerem Umfang die Maus. Neben Problemen der Grundlagenforschung bearbeiten wir zunehmend biomedizinische Projekte, in denen wir in Kollaboration mit klinischen Forscherinnen und Forschern Allele von Genen untersuchen, die beim Menschen Krankheiten verursachen. Ein Schwerpunkt dieser Arbeiten liegt auf der Untersuchung von Ciliopathien, also durch Ciliendefekte verursachte Krankheiten, die heute für sehr viele Erkrankungen verantwortlich gemacht werden und an denen jeder ca. 300. Mensch leiden soll.
Der Krallenfrosch Xenopus
Xenopus eignet sich in besonderer Weise für die Forschung an einer Universität:
- Embryonen sind durch künstliche Befruchtung in großer Zahl nach Bedarf verfügbar;
- die Embryonalentwicklung verläuft rasch, so dass instruktive Experimente in relativ kurzer Zeit durchführbar sind;
- Gene können durch ‚genome editing‘ (Stichwort CRISPR/Cas9) und antisinn Morpholino Oligomere (MO) manipuliert und ausgeschaltet werden. MOs können einseitig appliziert werden, so dass die nicht-manipulierte Seite als Kontrolle dient;
- Manipulationen können sehr zielgenau durchgeführt werden;
- die Haltungskosten sind im Vergleich zur Maus sehr viel günstiger;
- die Aussagekraft für den Menschen ist aufgrund der Konservierung der Genome hoch.

Die inneren Organe der Wirbeltiere sind asymmetrisch angeordnet: das Herz schlägt links (d.h. die Spitze zeigt nach links), der Blinddarm findet sich auf der rechten Seite, genau wie die Leber, Magen und Milz sind in der Bauchhöhle links zu finden, die Lungenflügel unterscheiden sich in der Zahl der Loben und der Darm windet sich asymmetrisch (Abb. 3). Diese Asymmetrie steht im Gegensatz zur ansonsten perfekten Spiegelsymmetrie des Körpers, mit paarigen Armen, Beinen, Augen, Ohren, Rippen usw. (Abb. 3). Für den gesunden Organismus ist die Organasymmetrie notwendig, z.B. für die Trennung von Lungen- und Körperkreislauf. Viele angeborene Missbildungen der Organe, vor allem des Herzens, gehen auf Fehler bei der Ausbildung der Asymmetrie im Verlauf der Embryonalentwicklung zurück.
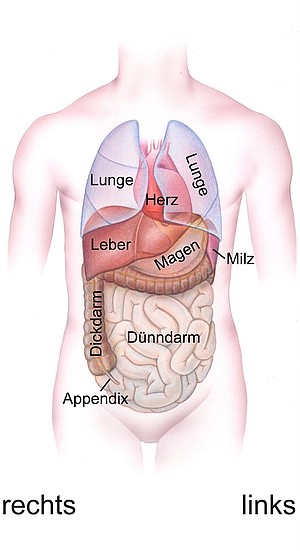
Abb. 3. Die Organasymmetrie des Menschen.
Die Gleichzeitigkeit von symmetrischer Entwicklung (z.B. des Skeletts) und asymmetrischer Ausprägung vieler Organe stellt den sich entwickelnden Embryo vor logistische Probleme. Die Aufklärung der zugrundeliegenden molekularen Mechanismen ist ein Schwerpunkt der Forschungsarbeiten im Labor von Prof. Blum und Prof. Schweickert, das heute zu den weltweit führenden auf diesem Gebiet zählt. Vor ca. 20 Jahren haben Blum und Schweickert, zeitgleich mit anderen Arbeitsgruppen, den Regelkreis entdeckt, der die Organasymmetrie im Wirbeltierembryo steuert. Die sog. Nodal-Signalkaskade wird vor der Entstehung der Organe im Embryo asymmetrisch, nur auf der linken Seite, aktiviert (Abb. 4). Der sekretierte Wachstumsfaktor Nodal induziert in Zielzellen drei Gene: seine eigene, die des Feedback-Inhibitors Lefty, der die Nodal-Aktivität zeitlich und räumlich begrenzt, und den Transkriptionsfaktor Pitx2, der die Asymmetrie der Organentwicklung entscheidend steuert.
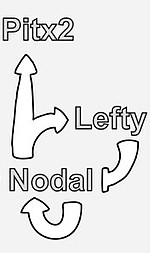
Abb. 4. Die asymmetrische Nodal-Kaskade im linken Seitenplattenmesoderm.
Eine entscheidende Frage im Zusammenhang mit der Nodal-Kaskade ist die links-asymmetrische Aktivierung in einem Embryo, der zu diesem Zeitpunkt perfekt bilateral-symmetrisch aussieht (Abb. 5).
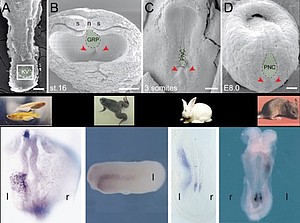
Abb. 5. Der Links-Rechts-Organisator (oben, grün eingefärbt) und links-asymmetrische Nodal mRNA Expression im linken Seitenplattenmesoderm (unten) in Fisch-, Frosch-, Kaninchen- und Mausembryonen (der Mausembryo ist in Bauchansicht gezeigt).
Forschungsarbeiten in unserem und anderen Laboren haben gezeigt, dass Cilien hierbei eine entscheidende Rolle spielen: im Links-Rechts-Organisator, der sich im Dach des frühembryonalen Darms befindet, rotieren Cilien, die entlang der Kopf-Schwanz-Achse ausgerichtet sind, im Urzeigersinn und erzeugen dabei einen nach links gerichteten Flüssigkeitsstrom im extrazellulären Raum (Abb.6 und Film 1).
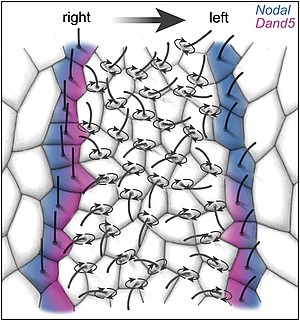
Abb. 6. Rotierende Cilien im Links-Rechts-Organisator erzeugen einen nach links gerichteten Flüssigkeitsstrom. Dieser unterdrückt den Nodal-Inhibitor Dand5 auf der linken Seite.
Bilden sich die Cilien nicht aus, sind sie unbeweglich oder nicht korrekt orientiert, kommt es unweigerlich zu Störungen der Nodal-Kaskade und – nachgeordnet – der Organasymmetrie. Viele menschliche Krankheitssyndrome, die auf Ciliendefekten beruhen (sog. Ciliopathien), gehen mit Störungen der Organasymmetrie einher.
Projekte
In zurzeit laufenden Projekten beschäftigen wir uns damit, wie der Flüssigkeitsstrom im Embryo wahrgenommen und die Nodal-Kaskade aktiviert wird, wie der Links-Rechts-Organisator entsteht und wie Asymmetrie im Verlauf der Evolution entstanden ist. Neben dem Südafrikanischen Krallenfrosch Xenopus untersuchen wir u.a. auch Seeigelembryonen. Teilaspekte aller Projekte können im Rahmen von Bachelor- und Masterarbeiten bearbeitet werden. Ansprechpartner für Interessenten ist Prof. Blum (martin.blum@uni-hohenheim.de).
Wir konnten in den letzten Jahren zeigen, dass als unmittelbarer Effekt des Flüssigkeitsstrom ein Inhibitor von Nodal, der Dand5 genannt wird, unwirksam wird. Der Verlust der Dand5 Aktivität findet nur linksseitig statt, so dass Nodal seine Aktivität auch nur dort entfalten kann (Abb. 6). Inzwischen wissen wir, dass hierfür micro-RNAs notwendig sind, neben einer Reihe weiterer Faktoren. Wir untersuchen im Frosch Xenopus, wie diese Regulation im Detail funktioniert und kooperieren dazu mit zwei Arbeitsgruppen in Japan (RIKEN, Kobe) und USA (Princeton), die diese Fragen in Maus und Zebrafisch verfolgen. Bearbeitet wird dieses Projekt von Markus Maerker, der über dieses Thema promoviert.
Wir wissen inzwischen, aus welchem Gewebe der Links-Rechts-Organisator entsteht. In zwei Projekten, die von den Doktorandinnen Sabrina Kurz und Melanie Tingler bearbeitet werden, untersuchen wir die daran beteiligten Signalwege. Im Links-Rechts-Organisator selbst gibt es Zellen, die mit beweglichen Cilien den Flüssigkeitsstrom erzeugen und solche, die diesen mit Hilfe von unbeweglichen, sensorischen Cilien wahrnehmen. Diese Zellen haben unterschiedliche Entwicklungsschicksale. Wie es zur Ausbildung dieser beiden Zelltypen kommt und welche Genprogramme sie unterscheiden, untersuchen wir im Krallenfrosch Xenopus.
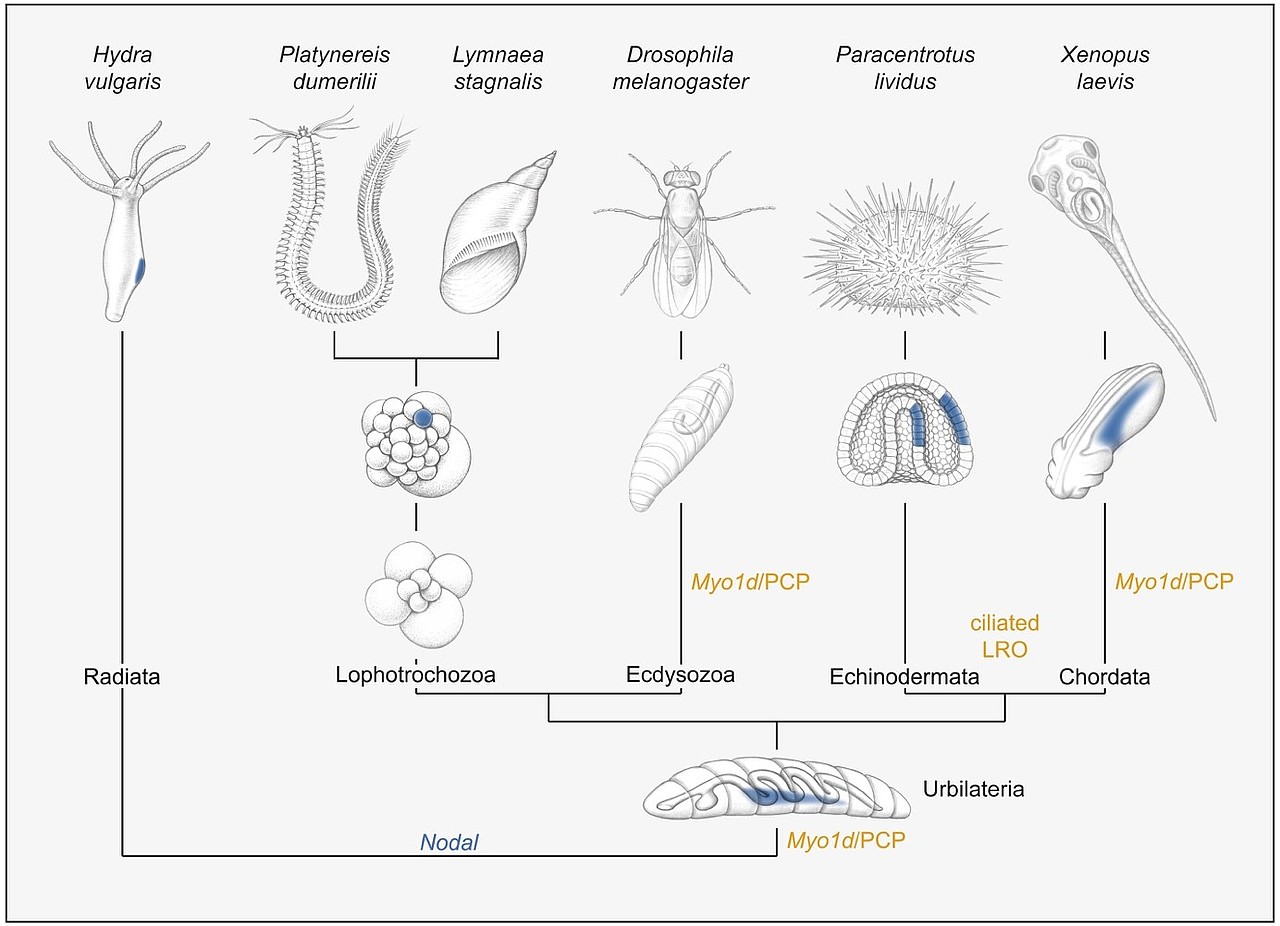
In einem laufenden Projekt, das von Melanie Tingler betreut wird, untersuchen wir ein Gen, myo1d, das für die asymmetrische Entwicklung des Darms und der Gonaden in der Taufliege Drosophila notwendig ist. Überraschenderweise wird dieses unkonventionelle Myosin auch im Krallenfrosch benötigt, u.a. für die Ausbildung des asymmetrischen Flüssigkeitsstroms. Dieser Befund spricht dafür, dass die grundlegenden Mechanismen von Organasymmetrie im Tierreich evolutionär konserviert sind, eine Hypothese, die durch frühere Arbeiten unserer Arbeitsgruppe, u.a. an Seeigel-, Kaninchen- und Schweineembryonen, gestützt werden (vgl. Abb. 7).
Goosecoid (Gsc) markiert in allen Wirbeltieren den sog. Spemann-Organisator. Dieses nach dem (in Stuttgart geborenen und begrabenen) Nobelpreisträger Hans Spemann benannte Gewebe ist für die Ausbildung der Körperachsen im Wirbeltierembryo verantwortlich. Wird dieses Gewebestück, die obere Urmundlippe, auf die gegenüberliegende Seite eines Empfängerembryos transplantiert, dann entwickelt sich ein siamesischer Zwillingsembryo. Dieses Organisatorexperiment, welches Hilde Mangold im Spemann-Labor in Freiburg 1924 durchgeführt hat, ist auch fast 100 Jahre nach der ersten Beschreibung molekular nicht vollständig aufgeklärt. Obwohl Gsc, wie die Transplantation der Urmundlippe, das Organisatorphänomen auslösen kann, entwickeln Mausembryonen auch nach Genverlust eine korrekte Körperachse, zeigen aber eine Vielzahl von Defekten, die während der späteren Entwicklung auftreten.
Wir konnten in den vergangenen Jahren zeigen, dass Gsc neben der Funktion im Spemannorganisator eine zweite Funktion hat bei der Ausbildung von planarer Zellpolarität, die durch Gsc inhibiert wird. Planare Zellpolarität spielt in vielen Prozessen im Embryo eine wichtige Rolle, so z.B. beim Schluss des Neuralrohrs. Interessanterweise ist die Funktion von Gsc nur bei Wirbeltieren zu finden. Das Gsc Gen der Taufliege Drosophila weist diese Funktion nicht auf.
Gsc als Regulator von planarer Zellpolarität
Nicole Henninger untersucht im Rahmen ihrer Doktorarbeit die Mechanismen der Gsc-abhängigen Repression planarer Zellpolarität. Dazu identifiziert sie die Bereiche des Proteins, die für diese Funktion notwendig sind, und spürt Proteine auf, die mit diesen Regionen interagieren. Unsere Hypothese geht davon aus, dass diese Funktion, die in den Wirbeltieren neu aufgetreten ist, für einige der Neurungen der Vertebraten wie Ausbildung des Schädels, Verhalten und Differenzierung von Neuralleistenzellen etc. eine wichtige Rolle gespielt haben könnte. Außerdem beschäftigt sich Nicole Henninger auch mit der Rolle von Gsc im Organisator selbst.
In der klinischen Forschung werden mit den modernen Methoden der Genomanalytik in rascher Folge neue Kandidatengene für humane Erkrankungen identifiziert. Ob diese tatsächlich krankheitsauslösend sind, kann nur in vivo, im Tiermodell geklärt werden. Dazu werden die entsprechenden (orthologen) Gene in Xenopus ausgeschaltet und mutante Embryonen und Kaulquappen auf Krankheitssymptome untersucht. Hierbei kommen Morpholino Oligomere und ‚genome editing‘ mit CRISPR/Cas9 zum Einsatz. Lässt sich im Froschembryo ein Defekt nachstellen, welcher der humanen Erkrankung entspricht, wird versucht, diesen Defekt durch Einbringen des gesunden humanen Gens zu korrigieren. Gelingt dies, kann anschließend das putativ krankheitsauslösende Gen in seiner Kapazität zum Retten des Defekts getestet werden. Aus diesen Untersuchungen lassen sich in aller Regel Erkenntnisse zur Ursache der Erkrankung ableiten. Das Tiermodell kann darüber hinaus zur Erprobung von neuen Therapieansätzen dienen.
Dieser Ansatz ist besonders für Ciliopathien erfolgversprechend, da die Froschlarve eine Vielzahl von Geweben mit beweglichen und sensorischen Cilien aufweist, z.B. in der Haut, wo hunderte Cilien auf multiciliierten Zellen Schleim vom Embryo weg transportieren, in dem sich im Wasser befindliche Partikel und Keime verfangen. In dieser Hinsicht entspricht die Haut der Larve dem Atemwegepithel der Säuger. Defekte offenbaren sich bereits beim Blick durch das Mikroskop, da sich die Embryos in diesen frühen Entwicklungsstadien nur durch die Bewegung dieser Cilien fortbewegen.
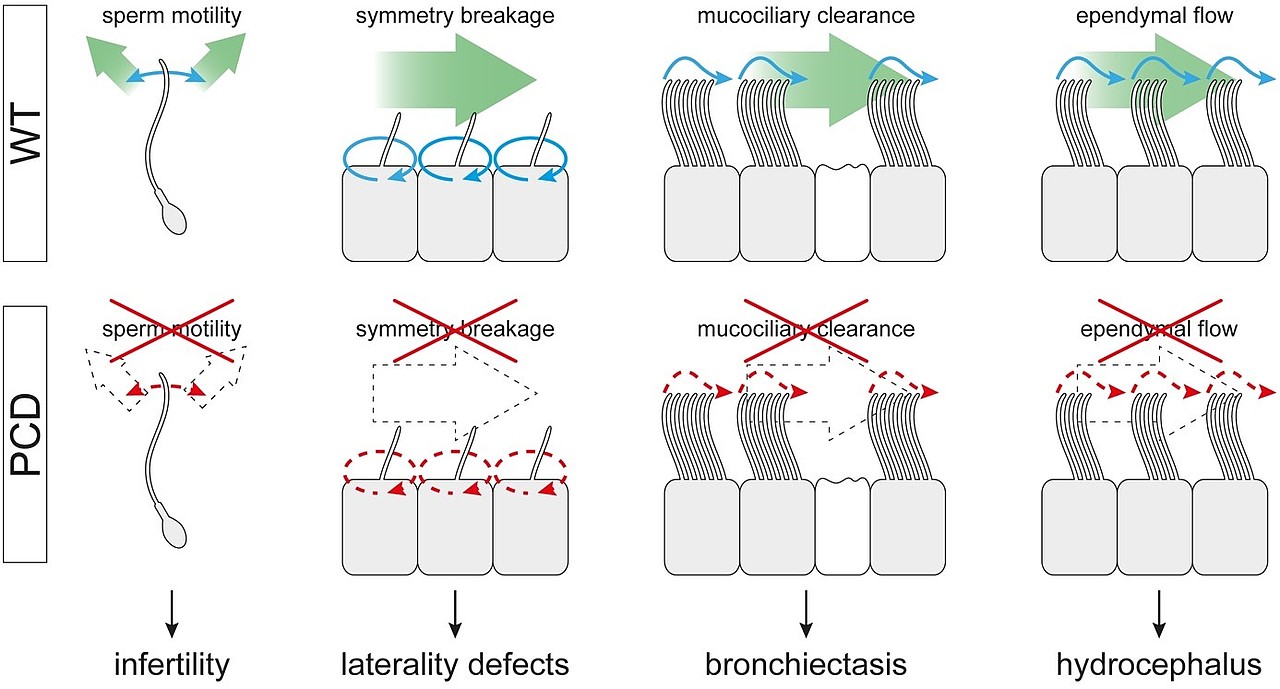
Unter primärer ciliärer Dyskinesie (PCD) versteht man Krankheitsbilder, die auf Fehlfunktionen von beweglichen Cilien beruhen. Unter PCD fallen z.B. Veränderung der Organasymmetrie und Unfruchtbarkeit aber auch eine Gruppe von Atemwegserkrankungen, die durch Ciliendefekte im Atemwegsepithel gekennzeichnet ist. Knapp 40 Gene sind heute bekannt, deren Mutation PCD auslösen kann. Damit lassen sich ca. zwei Drittel aller PCD-Fälle erklären. Viele weitere harren ihrer Identifizierung.

In Zusammenarbeit mit Prof. Achim Gossler von der Medizinischen Hochschule Hannover untersuchen wir eine Reihe von Genen, die im Verdacht stehen, PCD auslösen zu können. Diese Gene wurden von Prof. Gossler als Zielgene des Transkriptionsfaktors Foxj1 identifiziert, der das Programm der motilen Ciliogenese im Embryo steuert. Die AG Gossler untersucht diese Gene in der Maus, wir in Hohenheim die orthologen Gene im Krallenfrosch. Diese Untersuchungen, die bereits eine Reihe von Veröffentlichungen ergeben haben und die von Tim Ott und Franziska Fuhl im Rahmen ihrer Doktorarbeiten durchgeführt werden, haben eine Vielzahl von Ciliendefekten offengelegt. Interessanterweise ist das Spektrum der Defekte in Maus und Frosch nicht identisch, was darauf hinweist, dass Gendefekte in vivo funktionell kompensiert werden können.
Das Joubert Syndrom (JS) ist eine schwere Erbkrankheit, bei der Ciliendefekte im Gehirn u.a. zu motorischen und mentalen Störungen führen. In Zusammenarbeit mit Christina Evers und KollegInnen am Zentrum für Seltene Erkrankungen der Universität Heidelberg untersucht Tim Ott zwei neue Allele eines Kandidatengen, dass die Genetiker in Heidelberg bei einem betroffenen Kind identifiziert haben.
JS ist ein Beispiel einer Ciliopathie, die mit Lateralitätsdefekten kombiniert sein kann. Tim Ott untersucht im Rahmen seiner Doktorarbeit weitere Fälle, die durch fehlerhafte Anordnung der inneren Organe gekennzeichnet sind. Dazu kollaboriert er u.a. mit Bruno Reversade in Singapur, einem der führenden Labore bei der Aufklärung von Krankheitsmechanismen beim Menschen.
Serotonin (5-Hydroxy-Tryptophan, 5-HT) wird auf Grund seiner zentralnervösen Rolle als Neurotransmitter häufig als Glückshormon bezeichnet. Depressive Menschen zeigen in den entsprechenden Gehirnarealen häufig eine reduzierte neuronale Serotonin-Ausschüttung. Die bei weitem häufigsten eingesetzten Antidepressiva (SSRIs) erhöhen die 5-HT Konzentrationen und wirken daher Stimmungsaufhellend. Auch Migräne, Schizophrenie und Essstörungen zählen zu den Serotonin abhängigen mentalen Krankheiten. Weniger bekannt ist jedoch, dass im Körper die größte Menge an 5-HT nicht-neuronalen Ursprungs ist, sondern von spezialisierten Zellen des Darmepithels synthetisiert wird. Über das Blutsystem wird 5-HT im ganzen Körper verteilt und entfaltet dort zahlreiche physiologische Wirkungen, die eine medizinische Relevanz besitzen können.
Die zellspezifische 5-HT Aktivität wird über membranständige Rezeptoren vermittelt, die in großer Anzahl im menschlichen Genom codiert sind. Derzeit sind 14 Serotoninrezeptoren (5-HTR) beschrieben, die in 7 Familien zusammengefasst werden (5-HTR1-7) und bis auf eine Familie (5-HTR3) G-gekoppelte Rezeptoren darstellen.
Serotonin und dessen Rezeptoren werden auf Grund der medizinischen Bedeutung stark untersucht, was sich in derzeit über 140000 gelisteten Publikationen in Pubmed wiederspiegelt. Dennoch konnten wir, während der Frühentwicklung von Xenopus, eine bislang unbekannte Funktion für den 5-HT3 Rezeptor nachweisen. Xenopus 5-Htr3 ist verantwortlich für die Spezifikation des LRO Vorläufergewebes und dessen Funktionsverlust führte entsprechend zu Links-Rechts Achsendefekten. Überraschenderweise zeigte sich, dass die 5-HT3 Funktion dabei für die Aktivität des canonischen Wnt-Signalwegs notwendig war, welcher letztlich für die Zellschicksalsentscheidung verantwortlich war. Dieser Befund belegte erstmals eine enge Interaktion von Serotonin- und Wnt-Signaltransduktion, welche gerade in Bezug auf die Krebsforschung relevant sein könnte.
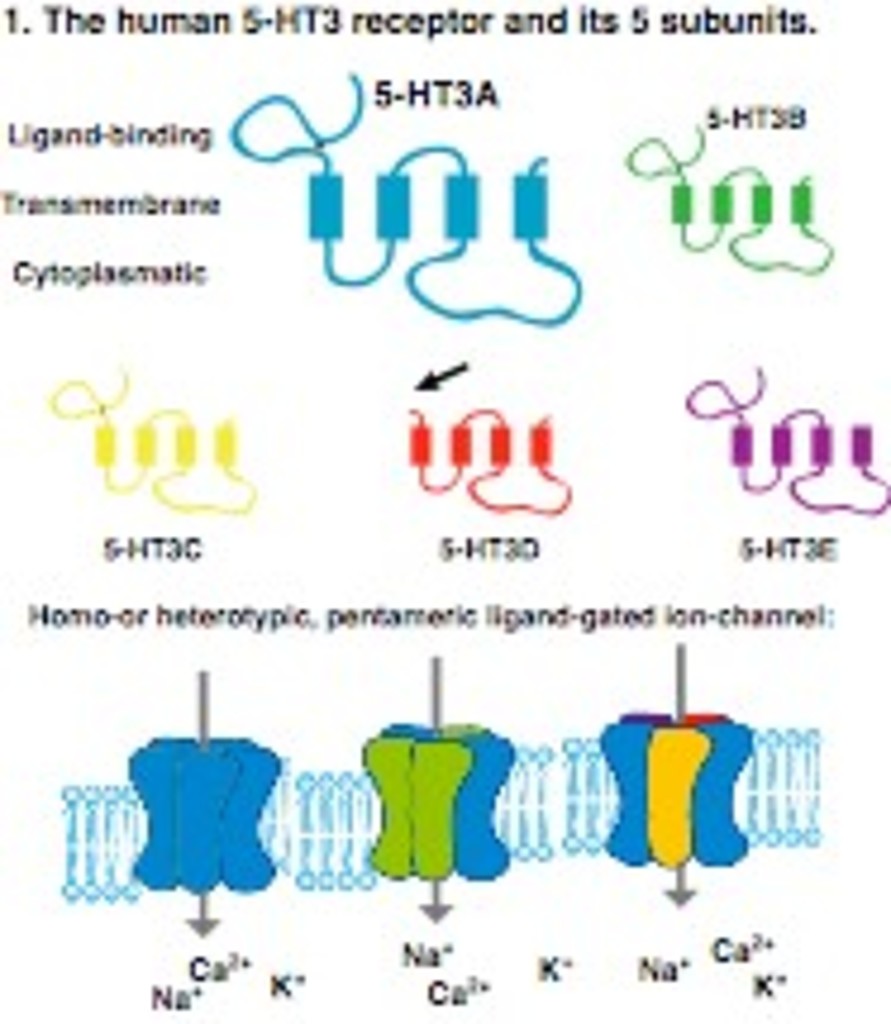
Der Serotoninrezeptor 3 (5-HT3R) gehört zu der Klasse von Liganden-gesteuerten Ionenkanälen. Das menschliche Genom enthält 5 Gene (HTR3A-E), welche Untereinheiten des Rezeptors kodieren (5-HT3A-E, Abb.X) die generell aus einer Liganden-Bindedomäne (LBD) und vier Transmembranregionen bestehen. Die 5-HT3D Untereinheit ist eine Ausnahme, da sie keine LBD aufweist. Interessanterweise fehlen Nagetieren (Mäuse, Ratten) und niederen Wirbeltieren die HTR3C-E Gene, was deren experimentelle Untersuchung erschwert. Der Rezeptor selber besteht aus 5 Untereinheiten, die variieren können. Die Untereinheiten zeigen ein breites Expressionsprofil im menschlichen Körper, besonders stark jedoch im Magen-Darm Trakt. Zahlreiche menschliche Krankheiten beruhen auf einer 5-HT3 Rezeptor Fehlfunktion wie etwa Depression und das Reizdarmsyndrom. (vgl. Abb.10.)
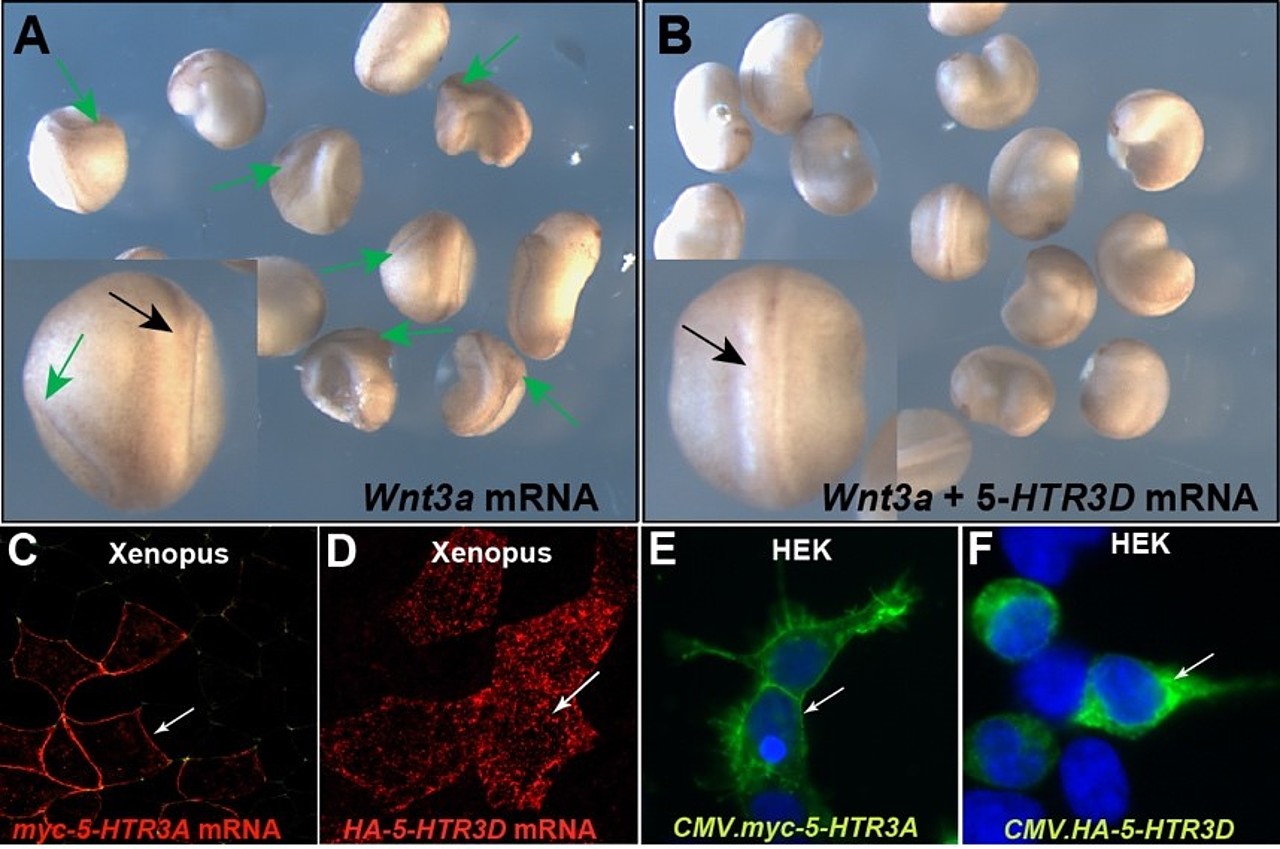
Um zu klären ob die menschlichen 5-HT3 Untereinheiten einen Einfluss auf den canonischen Wnt-Signal nehmen können, haben wir sie in einem heterologen Ansatz in Froschembryonen untersucht. Dafür verwendeten wir die Achsen-induzierende Potenz des Wnt-Signalwegs, die bei Fehlinduktion zu siamesischen Zwillingen führt und als Doppelachsen-Assay bezeichnet wird (Abb.ZZA). Überraschendweise zeigte sich, dass die Fehlexpression der 5-HTR3D Untereinheit die durch den Wnt3a-Liganden induzierten Achsen fast gänzlich hemmte (Abb. ZZB). Alle anderen Untereinheiten zeigten in diesem experimentellen Setup keine Wirkung. Auch in Zellkultur konnten wir den negativen Einfluss der 5-HT3D Untereinheit auf den Wnt-Signalweg nachweisen. In beiden Systemen Froschembryo und Zellkultur zeigten die 5-HT3A und 5-HT3D Untereinheiten die gleiche subzelluläre Lokalisation (Abb. ZZ C-F), was auf die Konserviertheit des Mechanismus hindeutet. Diesen haben wir bislang in 4 Master-Abschlussarbeiten untersucht und konnten zeigen, dass 5-HT3D negativ auf die Wnt-Liganden Sekretion einwirkt. Diese erstmalige beschriebene Funktion scheint unabhängig von der Ionenkanal-Aktivität zu sein und eröffnet daher völlig neue Forschungsansätze. Genauere Analysen der übrigen Untereinheiten sind sowohl im Frosch- als auch im Mensch-System geplant. (vgl. Abb.11.)
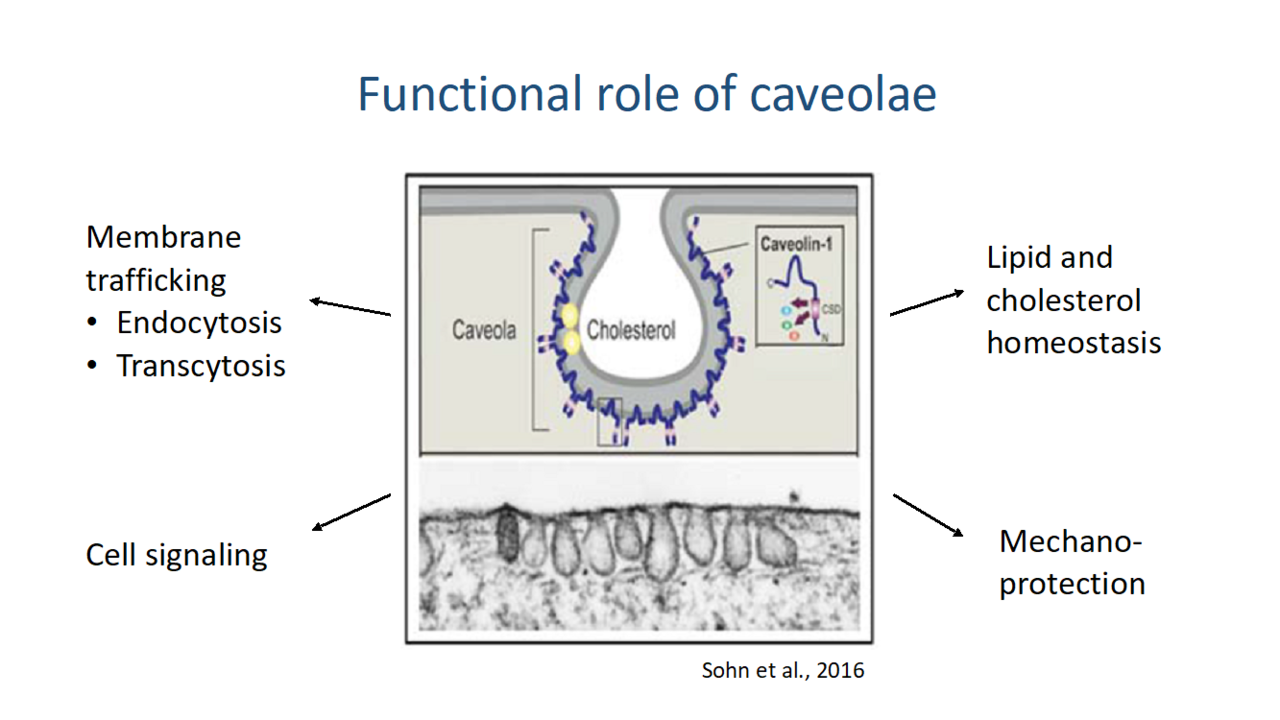
Caveolae sind kleine (50-80nm) Membraneinstülpungen, die in allen Zelltypen des Körpers vorkommen und eine Vielzahl von Funktionen besitzen. Neben Endozytose, Lipid-und Cholesterol-Homeostase werden Caveolae auch für Signaltransduktions-Prozesse verwendet (Abb.12.). Besonders spannend erscheint die Rolle von Caveolae bei dem Schutz der Plasmamembran vor mechanischem Stress, wie er beispielsweise bei Muskelzellen üblich ist. Bei Zug können sich Caveolae glätten und somit ein Reißen der Membran entgegenwirken. Proteine der Caveolin-Familie (Cav1-3) bilden das zentrale Rückgrat der Caveolae und können auch deren Bildung induzieren. Mutationen in den caveolin Genen führen zu Krankheiten mit unterschiedlichsten Ausprägungen. Caveolin3 wird besonders stark in Zellen der Muskulatur exprimiert, die besonders zahlreich Caveolae ausbilden. Caveolin3 Mutationen des Menschen führen zur Degeneration von Muskelzellen und letztlich zu deren Verlust.
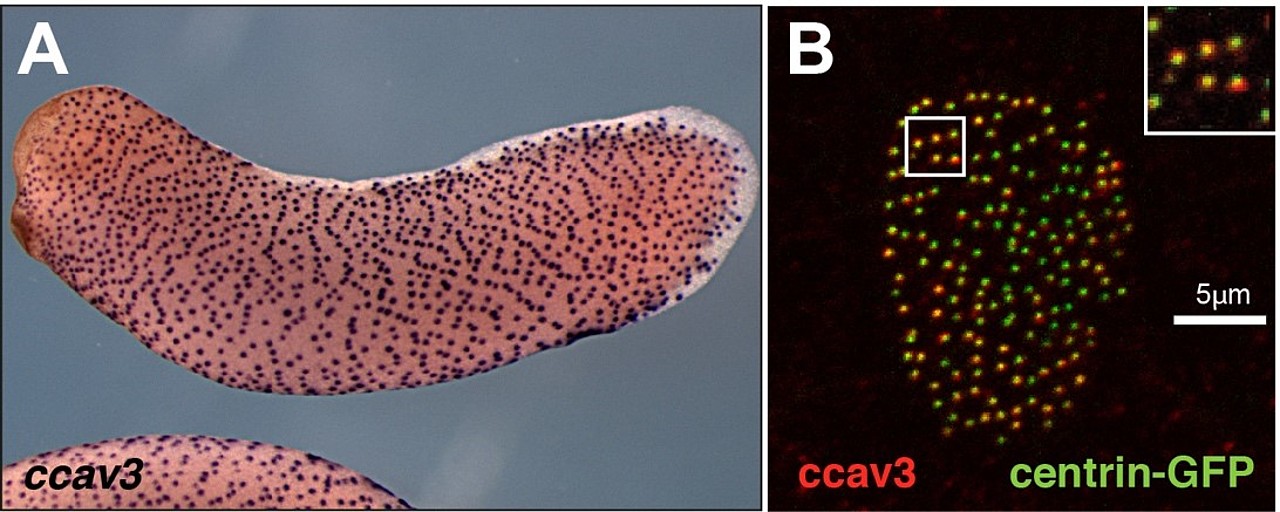
Wir konnten im Xenopus Genom drei Gen-Loci identifizieren, die Proteine homolog zum menschlichen Caveolin3 codieren. Eines dieser Xenopus cav3 Gene war überraschender ausschließlich in Geweben exprimiert, die durch multicilierte Zellen (MCCs) charakterisiert sind. Dieses cav3 Gen wurde daher als ciliäres cav3 (ccav3) benannt. (Abb.13.). Caveolae wurden bislang in MCCs nicht beschrieben. Der koordinierte Schlag von bis zu 300 Cilien pro Zelle übt jedoch einen mechanischen Stress auf die MCC Membran aus und daher könnten Caveolae als Schutz vor diesem dienen. Im Rahmen von zwei Abschlussarbeiten (Lehramt und Master) haben wir die ccav3 Funktion genauer angeschaut. Überraschenderweise fand sich ccav3 nicht in der Membran, sondern lokalisierte mit den Basalkörpern der Cilien (Abb.13), was gegen eine Caveolae-Funktion spricht. Der ccav3 Funktionsverlust störte zudem die MCC Ciliogenese, was eine komplett neue, bislang unbekannte Funktion für ein Caveolin darstellt. Zukünftig werden wir Signalwege bzw. Interaktionspartner identifizieren, über die ccav3 seine Funktion während der Ciliogenese vermittelt.